Представьте себе: вы вдруг слышите громкий топот копыт. Что первое приходит вам на ум? Лошадь, верно? Но почему именно лошадь, а не зебра?
Точно так же в медицине, когда перед нами встаёт задача постановки диагноза, мы часто идём по пути наименьшего сопротивления, выбирая наиболее очевидный диагноз. Однако, как и в случае с лошадью и зеброй, важно помнить о менее распространённых и, зачастую, более серьёзных состояниях .
В этой статье мы окунёмся в мир дифференциальной диагностики такого редкого заболевания как дефицит лизосомной кислой липазы (ДЛКЛ).
Сложность постановки диагноза ДЛКЛ заключается в том, что заболевание встречается крайне редко в практике врача
Поэтому очень часто о ДЛКЛ просто забывают или думают в последнюю очередь, а с учётом разнообразия клинических признаков пациент может долгое время наблюдаться с другим (неправильным) диагнозом. При этом ДЛКЛ может начать быстро прогрессировать в любом возрасте, поэтому очень важно вовремя заподозрить и поставить диагноз, чтобы начать терапию.
Для начала, вопрос: знаете ли вы что-нибудь о болезни Вольмана (БВ) или болезни накопления эфиров холестерина (БНЭХ)?


В настоящее время они признаны единым заболеванием и объединены под названием дефицит лизосомной кислой липазы или кратко — ДЛКЛ , которое характеризуется значительной тяжестью и преждевременной смертностью у пациентов всех возрастов.
Болезнь накопления эфиров холестерина — форма заболевания детей и взрослых старше 6 месяцев.
Болезнь Вольмана — это младенческая форма заболевания с дебютом в первые 6 месяцев жизни, которая быстро прогрессирует и, при отсутствии лечения приводит к летальному исходу в течение 1 года жизни.
Обратите внимание, что клинические проявления могут варьироваться в зависимости от возраста манифестации.
Истории пациентов

ДЛКЛ — редкое генетическое заболевание, которое связано с недостаточной активностью фермента — лизосомная кислая липаза (ЛКЛ).
активность фермента может составлять при инфантильной форме заболевания
Что скрывает в себе недостаточность фермента ЛКЛ?
ЛКЛ является ключевым ферментом, участвующим в расщеплении жиров внутри клеток организма, который локализуется в лизосомах, где происходит деградация макромолекул, включая липиды. При ДЛКЛ наблюдается снижение активности или полное отсутствие фермента ЛКЛ, что ведёт к накоплению эфиров холестерина и триглицеридов в тканях и органах по всему организму, что приводит к серьёзным последствиям .
Прогрессирующие последствия ДЛКЛ приводят к поражению жизненно важных органов и губительным последствиям*

Гепатомегалия, поражение печени, ↑АЛТ и ↑АСТ
Микровезикулярный или смешанный стеатоз, фиброз и/или цирроз печени, портальная гипертензия, печеночная недостаточность
* Bernstein DL, et al. J Hepatol. 2013;58(6):1230-1243; Reiner Ž, et al. Atherosclerosis. 2014;235(1):21-30; Radhakrishnan N. Splenomegaly.
http://misc.medscape.com/pi/android/medscapeapp/html/A206208-business.html. Дата обращения: 05.05.2024
Осложнения ДЛКЛ затрагивают жизненно важные органы и системы (печень, сердечно-сосудистую систему, селезёнку и желудочно-кишечный тракт) и очень часто носят полиорганный характер — более, чем у 85% пациентов . [3,4]
Почему возникает недостаточность фермента ЛКЛ?
ДЛКЛ это заболевание, которое передаётся по аутосомно-рецессивное типу наследования (то есть, когда оба родителя являются носителями заболевания). Недостаток фермента ЛКЛ возникает из-за мутации в гене LIPA.[5]
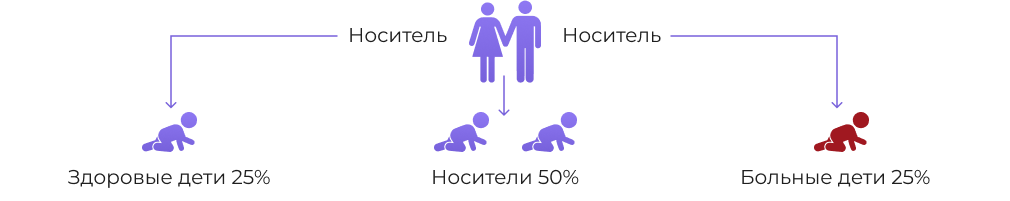
Ген LIPA картирован на хромосоме (10q23.31). При этом в более чем половине опубликованных случаев заболевания встречается патогенный аллель с синонимичной заменой, нарушающей сайт сплайсинга в экзоне 8 c.894G> A (E8SJM). [6]

Между генотипом и фенотипом корреляций не выявлено, но c.894G>A чаще встречается у детей старшего возраста и взрослых (не младенцев). [6]
Точные данные о распространённости ДЛКЛ неизвестны. Распространённость ДЛКЛ вариабельна в зависимости от факторов этнической принадлежности и географического положения. Считается, что частота ДЛКЛ составляет в среднем 1:40000–1:300000 живых новорождённых. [6]
Основные клинические проявления, на которые важно обращать внимание врачу-инфекционисту
Один из основных симптомов при ДЛКЛ — повреждение печени — поэтому вклад в диагностику ДЛКЛ каждого врача, который в своей практике сталкивается с большим количеством пациентов с повреждением печени, в частности, НЕинфекционной природы, очень большой.
Важно, чтобы на этапе диагностики пациентов проводилась комплексная оценка состояния для своевременного выявления редкого заболевания ДЛКЛ.
Если кратко говорить про симптомы, на которые стоит обращать внимание врачу-инфекционисту, то это:

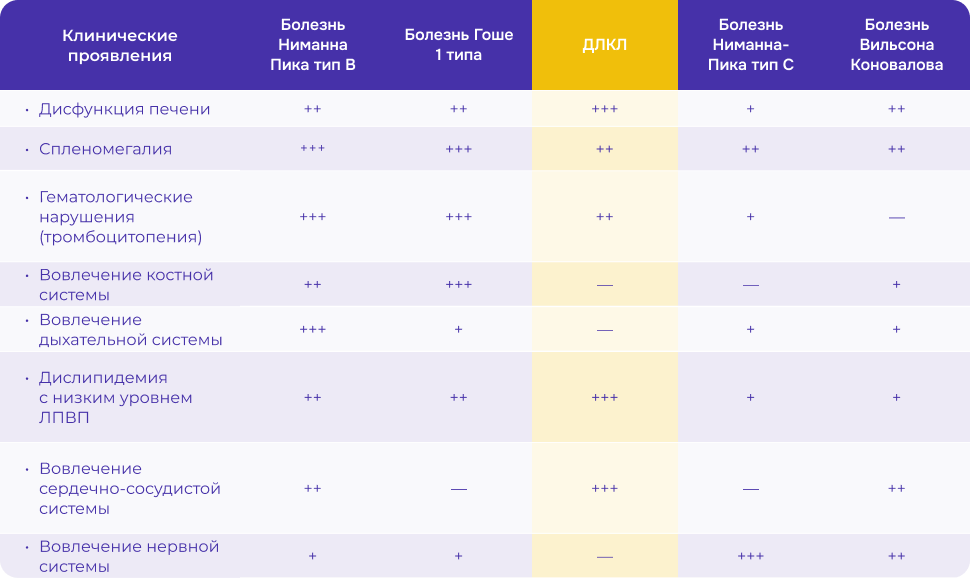
Клинические проявления ДЛКЛ [6]
- увеличение размеров печени
- увеличение размеров селезёнки
- увеличение размеров живота
- снижение массы тела
- задержку физического развития
- задержку психомоторного развития, гиперрефлексию
- желтушность кожных покровов
- признаки печёночной недостаточности
- отягощённый семейный анамнез (сходные симптомы у родных братьев и сестёр, близкородственный брак родителей)
- случаи внезапной детской смерти в семье
- гепатомегалию или гепатоспленомегалию
- диарею, боли в животе, стеаторею (при вовлеченности в патологический процесс кишечника)
- носовые кровотечения
- асцит
Также у пациентов в анамнезе может быть недавно перенесённая ЦМВ-инфекция или другие инфекции с сохраняющимся астеническим синдромом, длительные боли в животе, жалобы на боль в правом подреберье, могут наблюдаться признаки коагулопатии.
На что ещё стоит обратить внимание при сборе анамнеза и осмотре?
Стеатоз печени, гепатомегалия и повышенные трансаминазы являются ранними индикаторами накопления эфиров холестерина и триглицеридов в гепатоцитах и клетках Купфера. [3,8,9]
ПРИ ДЛКЛ наблюдаются высокие темпы прогрессирования поражений печени по сравнению с другими заболеваниями:
ДЛКЛ скрывается за масками разных заболеваний
Специалисты проекта подготовили для вашего удобства общую схему-алгоритм действий, которая поможет вам при подозрении на ДЛКЛ. [6]
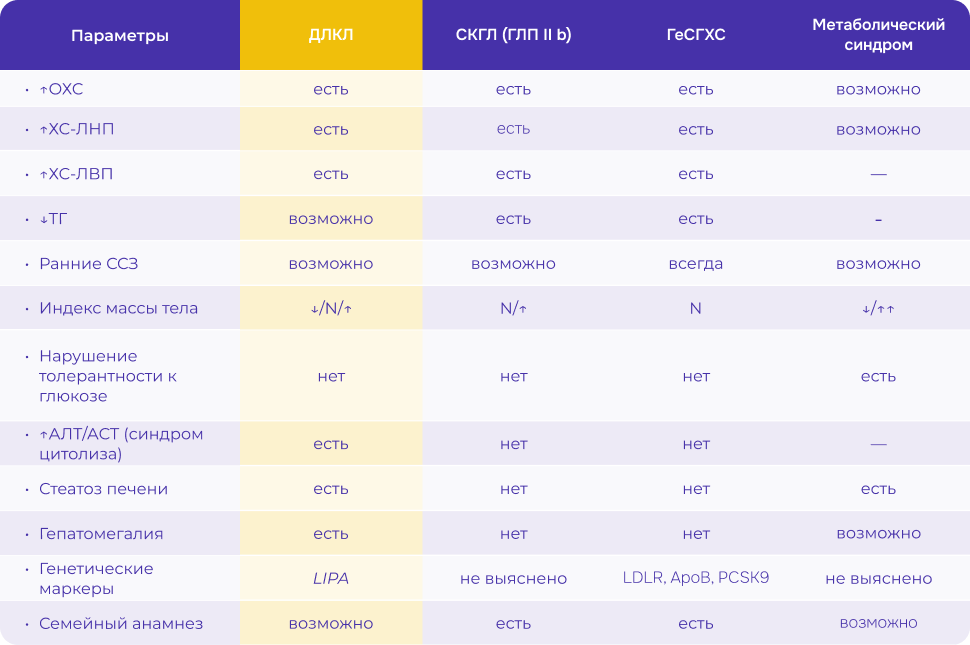
Важно проводить дифференциальную диагностику ДЛКЛ с другими заболеваниями печени и липидного обмена, а также — другими редкими болезнями:
- неалкогольной жировой болезнью печени (НАЖБП)
- неалкогольным стеатогепатитом (НАСГ)
- болезнью Ниманна-Пика тип А, В, С
- болезнью Гоше
- ганглиозидозами
- нарушениями обмена гликогена, жирных кислот
- болезнью Вильсона-Коновалова
- семейной гиперхолестеринемией

Кроме того, следует обратить внимание на пациентов, которые длительно наблюдаться с диагнозом гепатит неясной этиологии
Среди инфекционных заболеваний, в первую очередь дифференциальную диагностику необходимо проводить с герпесвирусными поражениями печени и другими вирусными инфекциями, такими как мононуклеоз.
Наиболее часто ДЛКЛ скрывается за маской НАЖБП или НАСГ
Ключевые клинические данные, позволяющие дифференцировать ДЛКЛ от НАЖБП/НАСГ включают: [3,10-12]
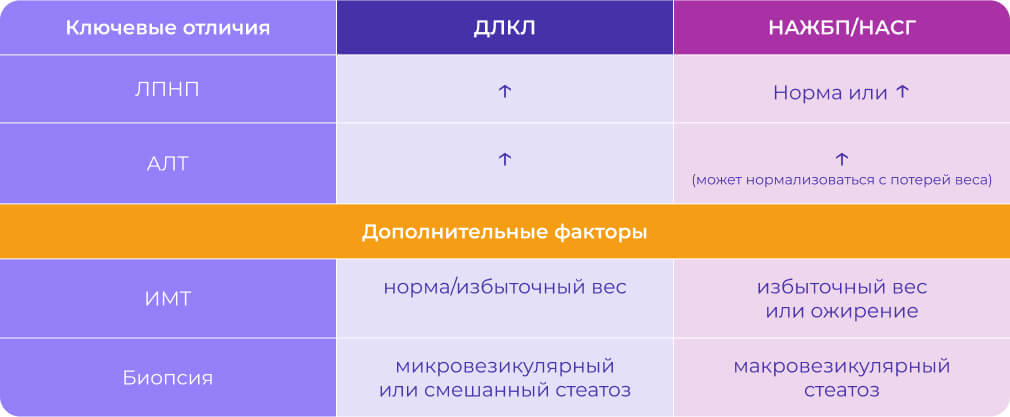
Если у пациента с подозрением на НАЖБП/НАСГ:
- уровень АЛт стойко повышен и не нормализуется с потерей веса и/или
- ЛПНП значительно повышены и/или
- Нет ожирения
- Быстро прогрессирует фиброз/цирроз

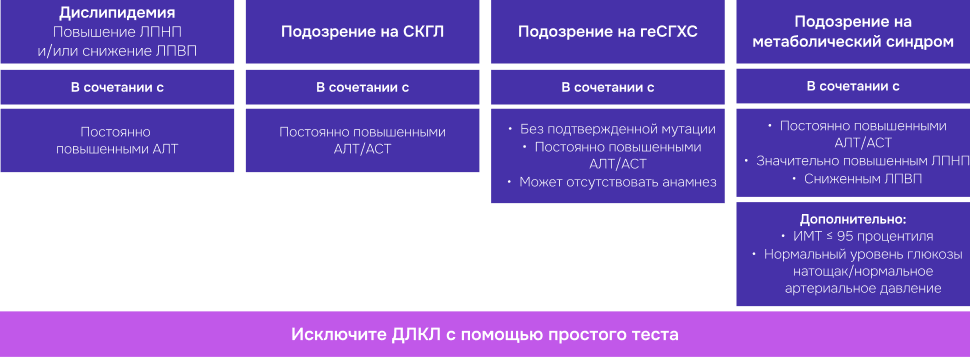


Лабораторная диагностика и маршрутизация пациента с подозрением на ДЛКЛ[3,18-22]
Ранняя диагностика важна для исключения или подтверждения любого «редкого» диагноза, чтобы вовремя начать необходимое лечение.
Для ДЛКЛ существует бесплатная и добровольная программа диагностики, которая проводится на базе лаборатории ООО «Гемотест» совместно с ФГБУ «Медико-генетического научного центра имени академика Н.П. Бочкова» и с ФГАУ «НМИЦ здоровья детей» , г. Москва. Программа осуществляется при поддержке ООО «Астразенека Фармасьютикалз» и не финансируется из средств ОМС.
В случае, если у вашего пациента до 18 лет наблюдается стабильное увеличение печени НЕинфекционной этиологии и нет подтверждённого диагноза, вы можете направить его на дополнительное обследование с целью исключения или подтверждения диагноза ДЛКЛ.
Какие анализы входят в программу




8 (800) 550-13-13 доб. 69777
Новые возможности для пациентов с ДЛКЛ
Как вы уже поняли, ДЛКЛ является редким генетическим заболеванием, требующим пристального внимания медицинского сообщества.
Несмотря на невысокую частоту встречаемости, оно способно приводить к серьёзным осложнениям и, в случае быстрого прогрессирующего течения, к преждевременной смерти. Раннее выявление и вовремя назначенная терапия имеют решающее значение для улучшения прогноза и повышения качества жизни пациентов.

Прогноз пациентов с ДЛКЛ

Важно, чтобы врачи разных специальностей были осведомлены о возможных симптомах ДЛКЛ и своевременно направляли пациентов на соответствующую диагностику. Это позволит не пропустить пациента с ДЛКЛ под масками других заболеваний или неустановленного диагноза и вовремя оказать такому пациенту эффективную медицинскую помощь, предотвратив дальнейшее прогрессирование.
Всего один простой тест позволяет исключить или подтвердить диагноз.
Заполнение формы врачом занимает всего одну минуту!
Получить форму электронного направления
Список литературы:
- Jones A., ValayannopoulosV., Eckert S at all. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med.2016 May;18(5):452–8.
- Burton BK, et al. Current Medical Research and Opinion, © 2017 Taylor Francis, reprinted by permission of the publisher Taylor & Francis Ltd., 33(7):1211–1214.
- Bernstein DL, et al. J Hepatol. 2013;58(6):1230–1243.
- Reiner Ž, et al. Atherosclerosis. 2014;235:21–30.
- LIPA (613497). Online Mendelian inheritance in man website. http://www.omim.org/entry/613497 Дата последнего обновления 06.02.2023. Дата обращения: 05.05.2024.
- Клинические рекомендации, Другие нарушения накопления липидов «Дефицитлизосомой кислой липазы», 2023.
- National Library of Medicine https://www.ncbi.nlm.nih.gov/clinvar/?term=LAL–D Дата обращения: 05.06.2024.
- Ambler GK, et al. JIMD Reports. 2012; 8:41–46.
- Ferry GD, et al. J Pediatr Gastroenterol Nutr. 1991;12:376–8.
- Burton BK et al. J Pediatr Gastroenterol Nutr. 2015;61(6):619–625.
- Preiss D et al. Clin Sci. 2008;115(5):141–150.
- Burton BK, et al. N Engl J Med. 2015;373(11):1010–1020.
- Nelson, RA, and Bremer, AA. Metab Syndr Relat Disord. 2010;8(1):1–14.
- Varghese M. Ann Pediatr Cardiol. 2014;7(2):107–117.
- Najam O, et al. Cardiol Ther. 2015;4:25–38.
- Gaddi A et al. Vasc Health Risk Manag.2007;3(6):877–886.
- Veerkamp MJ et al. Arterioscler Thromb Vasc Biol. 2002;22:274–282.
- Pericleous M et al. Lancet Gastroenterol Hepatol. 2017;2:670–679.
- Aguisanda F et al. Cur Chem Genom Transl Med.2017;11:1–18.
- Hamilton J et al. Clin Chim Acta.2012;16;413:1207–10.
- Строкова Т. и др. РМЖ. 2017 № 15.
- Маевская М. РЖГГК. 2016;3:41–51.
Информационный материал подготовлен при поддержке компании «АстраЗенека»
ООО «АстраЗенека Фармасьютикалз», 123112, город Москва, 1-й Красногвардейский проезд, дом 21, строение 1, этаж 30. Тел.: +7 (495) 799-56-99
Номер одобрения: RU-24140. Дата одобрения: 18.03.2025. Дата истечения: 17.03.2027